

Abstract
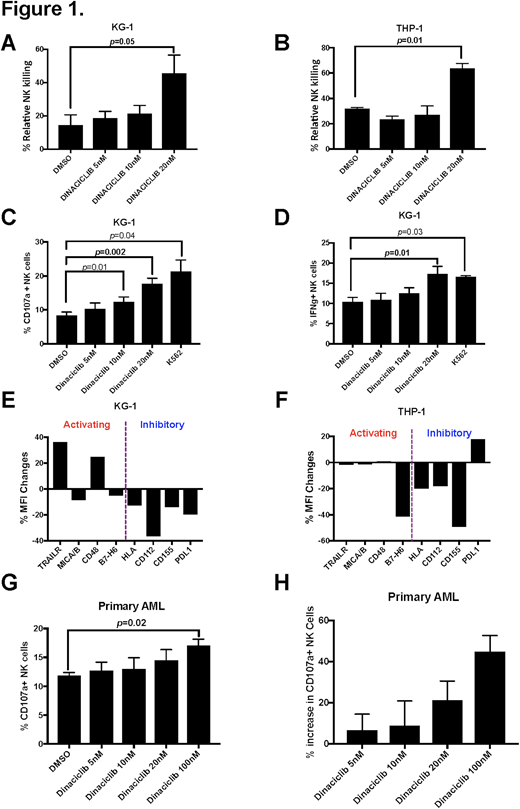
Dinaciclib, a CDK 1, 2, 5, and 9 inhibitor, has been shown to have potent anti-cancer activity against several malignancies. Our group previously demonstrated a direct RALB-dependent proapoptotic activity of dinaciclib against acute myeloid leukemia (AML) (Oncogene (2017) 36, 3263-3273). Furthermore, there is emerging evidence that dinaciclib can influence the recognition and killing of cancer cells by the immune system suggesting that it could have multimodal anti-cancer activities. As natural killer (NK) cells have a critical role in antineoplastic cytotoxicity, we hypothesized that dinaciclib may influence NK cell mediated cytotoxicity against human AML cells. To address this hypothesis, we evaluated the effects of dinaciclib on the NK cytotoxicity against human AML target cells and expression of known regulators of NK cell function. Specifically, we treated THP-1 and KG-1 cells with vehicle or dinaciclib at 5-20 nM for 24 hours, then washed the AML targets and added purified NK cells from PBMCs of healthy donors at an effector: target (E:T) ratio of 2:1. After 24 hours, viable AML target cells were enumerated using Fixable Aqua Dead Cell Stain and AccuCheck Counting Beads (Thermo Fisher Scientific) by flow cytometry. Relative NK cell target killing (%) was calculated by comparing the percentage of AML targets killed with drug treatment combined with donor NK cells relative to drug treatment alone in the absence of NK cells (Relative NK killing %). We assessed NK cell activation by measuring intracellular interferon-ɣ production and CD107a expression as well as NK ligand expression on dinaciclib-treated AML targets using flow cytometry. Dinaciclib treatment of AML targets enhanced the relative NK killing % for both KG-1 and THP-1 AML targets compared to control treatment (for KG-1, 45.6% vs 14.5%, p=0.05; for THP-1, 63.7% vs 31.7%, p=0.01) (Figure 1A, 1B) and was associated with enhanced NK cell degranulation as measured by CD107a expression (DMSO group, 8.3% vs dinaciclib 20nM group, 17.7%, p=0.002) and inflammatory cytokine production as measured by intracellular interferon-ɣ expression (DMSO group, 10.4% vs dinaciclib 20nM group 17.3%, p=0.01) (Figure 1C, 1D) supporting the ability of dinaciclib to sensitize AML targets for NK cell-based immunotherapy. To investigate potential mechanisms that promote NK cell activation by dinaciclib-treated AML targets, we assessed expression of several key NK cell ligands on AML targets and found that dinaciclib treatment led to decreased expression of inhibitory ligands including HLA class I, CD112, CD155 and increased expression in activating ligands such as TRAILR1, CD48 (Figure 1E, 1F) providing potential mechanistic insights. Notably, preliminary studies using a primary, patient-derived AML sample treated with dinaciclib resulted in a similar increase in the proportion of CD107a+ NK cells compared to the control group (44.7% increase, p=0.02, Figure 1G, 1H). While the detailed mechanisms for our findings remain to be determined, this is the first report to our knowledge that inhibiting CDK signaling can sensitize AML targets to NK cell based immunotherapy, a completely novel treatment approach. Further studies are ongoing to investigate the potential for combined targeted therapy with dinaciclib and adoptive NK cell therapy including patient derived xenograft (PDX) mice to validate its translational potential.
Felices:GT Biopharma: Research Funding.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal